
Gene detection and clinical characteristics analysis of cardiomyopathy in 32 children
-
摘要: 目的 探讨基因检测对儿童心肌病遗传病因的诊断价值,并分析其临床特征及基因型-表型的关系。方法 回顾性分析2018年1月—2020年12月收治的32例心肌病患儿临床资料及基因结果。根据患儿基因结果分为基因突变阳性(A组)17例和基因突变阴性(B组)15例,并对比两组首诊左心室射血分数(LVEF)、随诊1年结局(是否死亡)之间的差异。结果 基因突变阳性率为53.1%(17/32),其中新生突变约占35.3%(6/17),临床意义不明(VUS)者46.9%(15/32)。随诊1年死亡率为28.1%(9/32),其中A组47.1%(8/17),B组6.7%(1/15),差异有统计学意义(P=0.018)。9例死亡患儿的突变基因为GAA 4例、TTN 3例、LMNA和TAZ各1例,其中1例GAA(c.875A>G)基因的致病性目前被分类为VUS。32例心肌病患儿以腹胀、食欲欠佳等症状最常见,仅少部分(6.2%)患儿以晕厥、休克等症状就诊。32例患儿LVEF为(49.9±18.1)%,A组LVEF为(50.8±17)%,B组LVEF为(48.9±19.8)%,差异无统计学意义(P>0.05)。结论 儿童心肌病缺乏特异性临床表现。基因突变阳性患儿近期预后相对较差。特别是GAA、LMNA、TAZ、TTN基因变异相关心肌病患儿,大多近期预后不良。Abstract: Objective To investigate the diagnostic value of genetic testing for the genetic etiology of cardiomyopathy in children, and to analyze its clinical features and genotype-phenotype relationship.Methods The clinical data and genetic results of 32 children with cardiomyopathy admitted from January 2018 to December 2020 were retrospectively analyzed. According to the genetic results of the children, they were divided into gene mutation-positive group A(17 cases) and gene mutation-negative group B(15 cases), and compared the left ventricular ejection fraction(LVEF) at the first diagnosis and the 1-year follow-up outcome(death or not) between the two groups.Results The positive rate of gene mutation was 53.1%(17/32), of which de novo mutations accounted for about 35.3%(6/17). The percent of unknown clinical significance(VUS) were 46.9%(15/32). The mortality rate one-year follow-up was 28.1%(9/32), of which 47.1%(8/17) in group A and 6.7%(1/15) in group B, with a statistically significant difference(P=0.018). The mutated genes of the 9 dead cases were GAA of 4 cases, TTN of 3 cases, LMNA of 1 case and TAZ of 1 case. Among them, 1 case of GAA(c. 875A>G) gene was currently classified as VUS. Among the 32 children with cardiomyopathy, abdominal distension and poor appetite were the most common symptoms, and only a small number(6.2%) of children presented with syncope and shock. The LVEF of 32 children was(49.9±18.1)%. The LVEF of group A was(50.8±17)%, and the LVEF of group B was(48.9±19.8)%, with no significant difference(P>0.05).Conclusion Children with cardiomyopathy lack specific clinical manifestations. The short-term prognosis of children with positive gene mutation is relatively poor. Especially in children with cardiomyopathy associated with GAA, LMNA, TAZ, and TTN gene variants, most of them have poor short-term prognosis.
-
Key words:
- child /
- cardiomyopathy /
- inherited /
- genetic testing
-
-

图 1 死亡GAA基因突变阴性患儿及其父母的sanger测序
Figure 1. sanger sequencing of dead GAA gene mutation-negative children and their parents
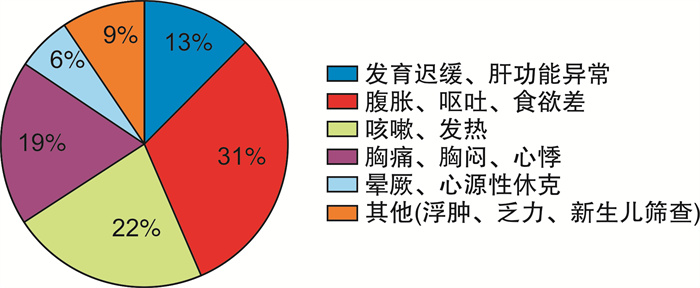
图 2 32例心肌病患儿入院时临床症状及体征分布图
Figure 2. Distribution chart of clinical symptoms and signs at admission in the 32 children with cardiomyopathy
表 1 17例基因突变阳性患儿的基因结果
Table 1. Genetic results of the 17 gene mutation-positive children
基因 性别 核苷酸 氨基酸 变异分类 变异来源 GAA 男 c.2853G>A
c.2853G>Ap.W851X
p.W851X疑似致病
疑似致病双亲 GAA 男 c.118C>T p.R40X 致病 父亲 MYH7 男 c.1357C>T p.R453C 致病 母亲 MYH7 男 c.2563_2565delGAG p.E855del 疑似致病 新生突变 MYH7 女 c.602T>C p.I201T 致病 母亲 GAA 女 c.1634C>T p.P545L 致病 母亲 GAA 女 c.1822C>T
c.1983G>Ap.R608X
p.W661X致病
致病双亲 LMNA 男 c.917T>G p.L306R 致病 新生突变 TTN 男 c.78938delA p.His26313Profs*2 疑似致病 母亲 TTN 女 c.5072_5074del p.1691_1692del 疑似致病 新生突变 TTN 男 c.4714C>T p.R1572X 疑似致病 父亲 FBN1 女 c.3596A>G p.D1199G 疑似致病 新生突变 TAZ 男 c.364_370+23delTGCCGAGGTGAGCTGCTCCTCC p.C122fs*15 疑似致病 母亲 BAG3 男 c.626C>T p.P209L 致病 新生突变 TNNI3 男 c.573G>A p.W191X 致病 新生突变 CTNNA3 男 c.1126C>T p.Q376X 疑似致病 母亲 SCN5A 男 c.3356G>A p.W1119X 疑似致病 父亲  下载: 导出CSV
下载: 导出CSV
表 2 17例基因突变阳性患儿临床资料及基因型-表型关系
Table 2. Clinical data and genotype-phenotype relationship of the 17 gene mutation-positive children
病例 首诊年龄/月 基因结果 首诊心脏超声结果 主要诊断 家族史 随访1年是否死亡 疾病/表型 首诊时LVEF/% 1 7 GAA 室间隔及左心室后壁增厚 HCM、GSDII 无 是 GSDII 46 2 3 GAA 左室及室间隔增厚 HCM、GSDII 无 是 GSDII 64 3 36 MYH7 室间隔、左室及右室部分心尖部增厚 HCM 无 否 DCM、HCM、LVNC 65 4 6 MYH7 室间隔、左室及右室心尖部增厚 HCM 无 否 DCM、HCM、LVNC 31 5 7 GAA 室间隔及左室壁肌层增厚,回声增强 HCM、GSDII 无 是 GSDII 52 6 2 GAA 室间隔及左室壁肌层及内膜增厚 HCM、GSDII 无 是 GSDII 73 7 54 MYH7 肥厚型心肌病、左室舒张功能减低 HCM 无 否 DCM、HCM、LVNC、Laing远端肌病 78 8 24 LMNA 全心大、部分心肌结构疏松、肺动脉高压 DCM 无 是 DCM 38 9 96 TTN 全心大、左心显著 DCM 无 是 DCM、HCM、ARVC 45 10 12 TTN 左心大、左心室部分心肌组织结构疏松 DCM 姐姐和父亲 是 DCM、salih肌病 29 11 11 CTNNA3 全心大、左室部分心肌组织疏松、增厚,肌小梁增大 DCM 无 否 ARVC、心肌病13型 27 12 84 FBN1 主动脉窦部增宽、左室增大 DCM 无 否 马凡综合征 38 13 8 TAZ 左心室大、心肌组织结构疏松,增厚,肌小梁粗大 LVNC 无 是 Barth综合征、DCM 70 14 156 TTN 左右室心肌组织结构疏松、增厚、肌小梁粗大 LVNC 姥姥 是 DCM、HCM、salih肌病 28 15 84 BAG3 双心房大、左室心包增厚、舒张功能减低 RCM 无 否 DCM 64 16 12 TNNI3 双心房扩张、双心室舒张功能减低 RCM 无 否 DCM、HCM、RCM 54 17 108 SCN5A 正常 长QT综合征 无 否 长QT综合征、DCM 62
下载: 导出CSV
表 3 15例基因检测阴性患儿的临床资料
Table 3. Clinical data of the 15 children with negative genetic testing
病例 首诊年龄/月 首诊心脏超声结果 家族史 随访1年是否死亡 临床诊断 首诊时LVEF/% 1 144 室间隔增厚、左侧冠状动脉、主肺动脉增宽 爷爷 否 HCM 83 2 8 室间隔及左室后壁增厚、心内膜回声增强 无 是 HCM 72 3 156 左心房、室间隔及部分左室间隔增厚 无 否 HCM 74 4 132 左心室大 无 否 DCM 66 5 6 左室大、左室内膜增厚 无 否 DCM 30 6 48 左室、房增大、左室壁厚度偏薄 无 否 DCM 30 7 108 左室稍大、左室心尖心肌结构稍疏松 无 否 DCM 63 8 120 左室稍增大、左室部分心肌结构疏松 无 否 DCM 63 9 96 左心大、左心室内膜增厚、心尖部心肌组织结构疏松 无 否 DCM 55 10 120 左心大、左心室内膜增厚、局部心肌组织结构疏松 无 否 DCM 29 11 7 左心大、左心室心尖部心肌组织结构疏松、增厚 祖母 否 DCM 42 12 7 左心室大、左心室心肌组织结构疏松、增厚、肌小梁粗大 无 否 LVNC 32 13 132 左心大,左、右室心尖部心肌组织结构稍疏松、肌小梁粗大 无 否 LVNC 25 14 24 左心室大、部分心肌组织结构疏松、肌小梁粗大 父亲和奶奶 否 LVNC 30 15 6 全心大、左室部分心肌组织疏松、增厚、肌小梁增大 无 否 LVNC 40
下载: 导出CSV
-
[1] 傅立军, 张浩. AHA儿童心肌病的分类和诊断科学声明解读[J]. 中国循环杂志, 2019, 34(S1): 49-53. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGXH2019S1011.htm
[2] De Angelis G, Bobbo M, Paldino A, et al. Cardiomyopathies in children: classification, diagnosis and treatment[J]. Curr Opin Organ Transplant, 2020, 25(3): 218-230. doi: 10.1097/MOT.0000000000000755
[3] Fadl S, Wåhlander H, Fall K, et al. The highest mortality rates in childhood dilated cardiomyopathy occur during the first year after diagnosis[J]. Acta Paediatr, 2018, 107(4): 672-677. doi: 10.1111/apa.14183
[4] Rupp S, Felimban M, Schänzer A, et al. Genetic basis of hypertrophic cardiomyopathy in children[J]. Clin Res Cardiol, 2019, 108(3): 282-289. doi: 10.1007/s00392-018-1354-8
[5] Orphanou N, Papatheodorou E, Anastasakis A. Dilated cardiomyopathy in the era of precision medicine: latest concepts and developments[J]. Heart Fail Rev, 2021, 14: 1-19.
[6] Ware SM, Wilkinson JD, Tariq M, et al. Genetic causes of cardiomyopathy in children: first results from the pediatric cardiomyopathy genes study[J]. J Am Heart Assoc, 2021, 10(9): e017731. doi: 10.1161/JAHA.120.017731
[7] 傅立军. 儿童心肌病的遗传学分子诊断[J]. 中华儿科杂志, 2021, 59(2): 158-160. doi: 10.3760/cma.j.cn112140-20201202-01070
[8] 中华医学会儿科学分会心血管学组儿童心肌病精准诊治协作组. 2006年至2018年国内33家医院4 981例住院儿童心肌病调查分析[J]. 中华实用儿科临床杂志, 2021, 36(13): 983-989. doi: 10.3760/cma.j.cn101070-20201108-01731
[9] 任媛媛, 张然然, 张丽, 等. GATA2缺陷2例报告及文献复习[J]. 临床血液学杂志, 2021, 34(1): 41-44. https://www.cnki.com.cn/Article/CJFDTOTAL-LCXZ202101010.htm
[10] Quiat D, Witkowski L, Zouk H, et al. Retrospective analysis of clinical genetic testing in pediatric primary dilated cardiomyopathy: testing outcomes and the effects of variant reclassification[J]. J Am Heart Assoc, 2020, 9(11): e016195. doi: 10.1161/JAHA.120.016195
[11] Xiao T, Zhou W. The third generation sequencing: the advanced approach to genetic diseases[J]. Transl Pediatr, 2020, 9(2): 163-173. doi: 10.21037/tp.2020.03.06
[12] 林丽容, 卢荔红, 胡雪群, 等. 家族性肥厚型心肌病MYBPC3基因变异及其临床表型分析[J]. 临床心血管病杂志, 2021, 37(6): 557-560. https://www.cnki.com.cn/Article/CJFDTOTAL-LCXB202106013.htm
[13] Norrish G, Kaski JP. The risk of sudden death in children with hypertrophic cardiomyopathy[J]. Heart Fail Clin, 2022, 18(1): 9-18. doi: 10.1016/j.hfc.2021.07.012
[14] Viamonte MA, Filipp SL, Zaidi Z, et al. Phenotypic implications of pathogenic variant types in Pompe disease[J]. J Hum Genet, 2021, 66(11): 1089-1099. doi: 10.1038/s10038-021-00935-9
[15] Wang Y, Han B, Fan Y, et al. Next-generation sequencing reveals novel genetic variants for dilated cardiomyopathy in pediatric chinese patients[J]. Pediatr Cardiol, 2022, 43(1): 110-120. doi: 10.1007/s00246-021-02698-8
[16] Ellepola CD, Knight LM, Fischbach P, et al. Genetic testing in pediatric cardiomyopathy[J]. Pediatr Cardiol, 2018, 39(3): 491-500. doi: 10.1007/s00246-017-1779-2
[17] Wilde AAM, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy[J]. JACC Clin Electrophysiol, 2018, 4(5): 569-579. doi: 10.1016/j.jacep.2018.03.006
[18] Herkert JC, Abbott KM, Birnie E, et al. Toward an effective exome-based genetic testing strategy in pediatric dilated cardiomyopathy[J]. Genet Med, 2018, 20(11): 1374-1386. doi: 10.1038/gim.2018.9
-
图(2)
表(3)
计量
- 文章访问数: 2330
- PDF下载数: 1279
- 施引文献: 0